非晶态高聚物从玻璃态到橡胶态,有一个转变——玻璃化转变。这个转变一般其温度区间不超过几度。但在转变前后,模量的减少达三个数量级。在实用上是从硬而脆的固体变成韧性的橡胶。所以,玻璃化转变是高聚物一个重要的特性。
形成玻璃态的主要原因,可能是高聚物分子结构不对称,不能形成结晶;也可能是没有足够的能量去重排结晶。而且多数高聚物也只有在特定的条件下方能结晶。同时高聚物很难形成100%的结晶,总有部分非晶态存在,因此玻璃化转变是高聚物普遍现象,只不过非晶态少的高聚物玻璃化转变不明显。
一、玻璃化转变温度的测定
高聚物在玻璃化转变时,除了力学性质有很大变化,其他性质如体积,热力学性质,磁性质等,都有很大变化。在理论上后面的变化更为重要。下面就简要介绍:
1、体积的变化
用膨胀计测定玻璃化温度是最常用的方法。一般是测定高聚物的比体积对温度的关系.把曲线两端的直线部分外推至交点作为Tg(如图1)

从图可以看出,玻璃化转变同冷却速率有关:冷却的快。得出的Tg高;冷却的慢,Tg就较低。同样,加热速率或快或慢,Tg也或高或低。产生这种现象的原因是体系没有达到平衡。但要达到平衡,需要很长的时间(无限长),这在实验上做不到。通常采用的标准是每分钟3℃。测量时常把试样在封闭体系中加热或冷却,体积的变化通过填充液体的液面升降而读出、这种液体不能和高聚物发生反应或溶解、溶胀,zui常用的是水银、也有人用空气作测量的流体,达时可测定压力的变化。
其它与体积有关的性质也可用于测定,加试样的折射系数、X射线的吸收等。
2、热力学方法
量热方法也是测定玻璃化温度的常用方法。在Tg时,热焓有明显变化,热容有—个突变。自从有了差热分析(DTA)和差示扫描量热计后,量热方法变得更为重要。
象体积变化一样,热焓和热容的变化也和速率有关:图2表示比体积(V)和焓(H)对温度的关系,图3表示体膨胀系数和热容对温度的关系,都出现行“滞后”现象。图中曲线1是缓慢冷却,曲线2是正常冷却和升温,曲线3是快速冷却;曲线1、3是正常升温。
3、核磁共振法(NMR)
利用电磁性质的变化研究高聚物玻璃化转变的方法是核磁共振法(NMR)。
在分子运动开始前,分子中的质子处于各种不同的状态,因而反映质子状态的NMR谱线很宽。当湿度升高,分子运动加速后,质子的环境被平均化,共振谱线变窄,到了Tg时谱线的宽度有了很大改变。图5给出了聚氯乙烯的NNR线宽(ΔH)的变化。由图5可得Tg为82℃。


除上述方法外,这里再列举几种测定玻璃化转变的方法,如图6、7、8所示。图6为甲基丙烯酸丙酯的折光率—温度曲线。由图6可见,T> Tg,dn/dT增加,即链段开始运动后光线在聚合物中的传播速度增加;图7为天然橡胶导热系数——温度曲线。可以看出,T>Tg时,链段的无规热运动阻碍了热能的定向传导,使λ值急剧减小;图8为聚酯酸乙烯酯的膨胀率——温度曲线。说明了T>Tg时,由于链段运动,“自由空间”增加,则dv/dT值急剧增大。

上面的这些测量方法多以玻璃化过程中发生的物理现象为基础。以不同物理量为准则的测量方法,所得玻璃化温度各不相同,而且试验测量也相当费力和耗时,而从自由体积理论、热力学理论和动力学理论来计算玻璃化温度尚存在许多问题。因此,有人另辟蹊径对聚合物的玻璃化温度作合理的推测。下面以硬脂酸乙烯酯+ 乙酸乙烯酯+ 氯乙烯体系形成的三元聚合物为例,采用神经网络法,建立了进料组成中各组分的摩尔分数、链节重量分数和聚合度等与玻璃化温度的定量映射关系,进而对未知条件下的三元聚合物玻璃化温度作出了预测。
神经网络作为一种并行仿生信息处理系统,由若干层神经元组成,各层神经元之间通过联接权进行信息传递与交换。本文采用具有三层结构的逆传播神经网络模型,网络拓扑结
构如图1 所示。各层神经元之间的信息通过SIGMOID 函数进行传递,以SDEC 为迭代收敛判据,以SDEP 表达网络的预测能力[3 ] 。二者定义如下:

式中SQRT 表示取平方根,i 和j 分别为训练集和测试集中的样本序号,加和分别遍及训练集和测试集所有样本点,yexp、ycal和ypred分别为实验值、标定值和预测值。网络的权值调整按如下方式进行wij = wij +αδi x j +ηΔw ij其中w ij为上一层中神经元i 到下一层神经元j 的联接权, x j为相应神经元的输出值,δ为对应神经元的误差项,ΔWij为上次迭代权值与前次迭代权值之差,a和η分别称为学习效率和冲量因子。有关神经网络逆传播算法的详细内容参见文献[4 ,5 ]。玻璃化温度标志着聚合物物理性质的巨大变化。在Tg 以上,玻璃态聚合物将变得柔顺而趋向于流动。要对Tg 做出预测,首先要了解影响Tg 的因素。在相同的试验条件下,影响聚合物Tg 的主要因素是聚合物的结构特性。包括链的柔性、支化情况、链规整性和聚合物的分子量。对于聚合物来说,不同成分的链节比例及各组成链节的重量分数都将影响Tg 值。链的柔性取决于组成主链的化学结构的本性,化学链易旋转的柔性链趋向于有低的Tg,若某种集团的引入阻碍链的旋转而使其僵硬,则会使Tg 升高,侧链的生成将阻碍链的旋转,从而提高Tg 值,侧链越大,这种效应越明显。另一方面,侧链的增大,降低了聚合物链间作用力,弱化了分子间力对Tg 的影响,侧链效应具有双重作用。从聚合物的链节成分比例看,各组成成分的差异,影响了链的规整性,规整性好,利于分子的敛集,限制了分子的内旋转,使玻璃化温度Tg 升高。从分子量效应看,随聚合物分子量的增加,端基分率减小,自由体积减小,链的自由度也减小,Tg 值则升高。

综合上述分析,在处理硬脂酸乙烯酯+ 乙酸乙烯酯+ 氯乙烯三元共聚体系时,我们选择了合成聚合物时进料组成中三个成分的摩尔分数(X1 ,X2 ,X3) ,以此表达聚合物组成链节比例的数量效应,考虑到各单体重量上的差异,我们选择了聚合物中硬脂酸乙烯酯的重量分数和乙酸乙烯酯的重量分数来表示链节组成上的重量效应(W1 ,W2) 链的柔性、规整性及支化情况均与此组成效应相关,比如当硬脂酸乙烯酯的重量分数超过约0. 4 时,聚合物链出现支化。对于分子量效应,我们选择了聚合度(N) 来表达。这样我们得到6 个影响Tg 的因素,由此构成6 维空间R6 (X1 ,X2 ,X3 ,W1 ,W2 ,N) ,而聚合物的Tg 将构成一维空间R1 ( Tg) ,为建立两个空间的联系,我们选择神经网络,采用631 网络拓扑结构,即输入层设置6 个节点,分别对应于6 个影响因素,输出层设置1 个节点,对应于Tg 的响应值,隐含层设置3 个节点,以适应由6 维空间到1 维空间映射的复杂性要求。训练过程的学习效率取为0. 35 ,冲量因子取为0. 85 。从文献中收集到的数据如表1 所示,取其中前36 组数据作为训练集,其余12 组数据为测试集。以训练集数据训练神经网络,构造由6 维空间到1 维空间的映射关系,当SDEC 达到4. 6915时,网络迭代8800 次,此时停止训练并转入对测试集的预测,以检验训练模型的预测能力,得到的SDEP值为3. 2253 。神经网络训练过程中误差演化曲线如图9 所示,实验值和预测值之间关系如图10 所示。从图9 可以看出,训练过程是收敛的,说明学习效率和冲量因子的选择是合理的。从图10 可以看出,实验值和预测值是比较接近的,说明由神经网络得到的模型是可靠的,实现的由6 维空间到1维空间的映射关系是正确的。

二、玻璃化转变的理论
玻璃化转变有很多理论,但不外乎从热力学的角度去计算理想玻璃态的熵,或是从玻璃化转变的松弛现象去考虑动力学过程。
1、自由体积理论
自由体积理论认为高聚物的体积是有两部分组成的,一部分是大分子本身的占有体积,另一部分是分子间的空隙,后者即为自由体积。在温度比较高时,自由体积较大,能够发生链段的短程扩散运动,而不断地进行构象重排。温度降低,自由体积减小,降至Tg以下时,自由体积减小到一临界值一下,此时链段的短程扩散运动已不能发生,高聚物表现为固体的性质,这是就发生了玻璃化转变。
WLF方程预示了玻璃化转变的等自由体积状态。根据WLF方程

根据Doolittle经验式,并假定在Tg时,自由体积分数为fg ;在Tg以上时,自由体积分数随温度线性变化。
由此可推导出WLF方程式的类似形式
于是,C1=B/2.303fg,C2=。B是Doolittle方程中的常数,近似等于1。
从许多实验事实,发现在大部分非晶态高聚物中,

由此,这就是说,玻璃化转变时高聚物的自由体积都等于体积的2.5%。上述结论可以用图12加以具体说明。Fox和Flory认为:在Tg时,玻璃态的比体积是Vg。其中部分是大分子的占由体积Vo’。Vo是绝对零度时的占有体积,随温度上升,分子热运动振幅增大,引起占有体积膨胀,替膨胀系数是βg。Vg中的另一部分是自由体积Vf,在Tg以下,分子运动冻结,Vf被冻结在高聚物中不再变化。于是,T=Tg时,比体积是

而T>Tg时,比体积是

式中βr是Tg以上的替膨胀系数,这时,除了占有体积的正常膨胀外,自由有体积也在膨胀,

从图12可以知道,自幼体积膨胀系数就是Δβ,Δβ=βr-βg=βf。当温度降到Tg时,自由体积的分数达到临界值,等于2.5%。这时,高聚物进入玻璃态。
自由体积理论是一个玻璃化转变处于一个等自由体机状态的理论,谈随着冷却速率不同,高聚物的Tg并不一样,因此Tg时的自由体机并不相同,同时,自由体积理论认为Tg以下自由体积不变,实际是会变得。Kovacs曾对高聚物的体积松弛做过大量的研究工作,他把淬火后的高聚物恒温放置,发现高聚物的体积随着放置时间的变长而不断变小,这表明自由体积在不断减小,但减小的速率越来越慢。高聚物中自由体的多少和其物性关系很大。
按照Ehrenfedst对平衡热力学的定义,转变可以分成一级转变和二级转变。如转变前后的两种物态分别用下标1、2表示,则在转变点两种物态的Gibbs自由能应相等,即

但在转变点对一级相变来说,自由能对温度T、压力P的一阶导数在转变时是不连续的;而对二级转变来说,二阶导数是不连续的;
一级转变

二级转变

根据热力学的关系,可以明确自由能F对T,P的一阶导数和二阶导数的物理意义。因为

其中S熵,V是体积,Cp是热容,β是体膨胀系数,κ是压缩系数。
因此,一级相变,在转折点,两种物态的熵和体积不相等。

二级转变,在转变点,两种物态的Cp,β,κ不相等

在玻璃化转变时,高聚物的Cp,β,κ恰恰都有不连续性,所以通常把玻璃化转变看作是二级相变,其实并没达到热力学平衡,因而不是真正的二级相变。
Gibbs和Dimarzio的理论,是玻璃化转变的热力学理论zui严密的代表,它通过对构象熵随温度的变化进行了复杂的数学处理,证明通过T2(高聚物熵为零真正二级转变时的温度)时,F和S是连续变化的,内能和体积也是连续变化的,但Cp和α不连续变化,从而从理论上预言,在T2时存在真正的热力学二级相变。
G—D理论认为,尽管事实上无法达到T2,因而无法用实验证明其存在。但是,在正常动力学条件下观察到的实验的玻璃化转变行为和T2处的二级转变非常相似,T2和Tg是彼此相关的,影响它们的因素应该相互平行,因此,理论得到了关于T2的结果,应当也适用于Tg。在这样的框架内,得到了一系列结果,很好的说明了玻璃化转变行为与交联密度,增塑,共聚和分子量的关系,也解释了压力对T2,Tg的影响。
G—D理论认为T2时存在真正的二级热力学转变,因此压力对Tg的影响可以直接从平衡热力学关系式求出。对于—级相转变,转变温度的压力依赖性由clapeyron方程确定

此式不能直接应用于二级转变,因为此时ΔV和ΔS都为零,dT/dP不确定。但是,我们可以援引L’Hopital法则,将式(13)右边的分子和分母分别求导,以求得极限值。将式
(13)右边的分子和分母分别对T求导,并根据式(12)可得

如果对压力求导,则结果为

所以压力对T2的影响,当然也就是对的影响,可以用式(14)和(15)表示。式
(15)和自由体积理论的结果式相同,并与实验结果基本相符,说明G—D理论是成功的。
G—D理论预言的热力学二级转变温度T2,可以由WLF方程求出。当取Tg作为参考温度时

式中。称为移动因子,是温度为时的松弛时间,C1和C2两个经验常数此时可取近似普适值.分别等于17.44和51.6。根据前面的分析,到T= T2时,构象重排需要无限长的时间,即;或者说,为将一个有着无限时间标尺的实验数据,移动到有限的时间标尺,必须取。显然,满足上述条件,方程式(16)右边的分子维持有限值时,分母必须变为零,即
因而
3、动力学理论
玻璃化转变现象有着明显的动力学性质,Tg与实验时间标尺有关,因此有人认为玻璃化转变是由动力学方面的因素引起的。
zui初的动力学理论认为,当高聚物收缩时,体积收缩有两部分组成:一是连锻的运动降低,另一部分是连锻的构象重排成能量较低状态,后者又一个松弛时间。在降温过程中,当构想重排的松弛时间适应不了降温速度,这种运动就被冻结出现玻璃化转变。
动力学理论的另一类型式位垒理论,这些理论认为大分子构想重排时涉及到主链上单键的旋转,键在旋转时存在位垒,当温度在Tg以上时,分子运动有足够的能量去克服位垒,达到平衡。但当温度降低时,分子热运动的能量不足以克服位垒,于是便发生了分子运动的冻结。
由于玻璃化转变是与分子运动有关的现象,而分子运动又和分子结构有着密切关系,所以分子链的柔顺性、分子间作用力以及共聚、共混、增塑等都是影响高聚物Tg的重要内因。此外,外界条件如作用力、作用力速率,升(阵)温速度等也是值得注意的影响因索。
分子链的柔顺性是决定高聚物Tg的最重要的因素。主链柔顺性越好,玻璃化温度越低。
主链由饱和单键构成的高聚物,因为分子链可以固定单键进行内旋转,所以Tg都不高,特别是没有极性侧基取代时,其Tg更低。不同的单键中,内旋转位垒较小的,Tg较低。例如:
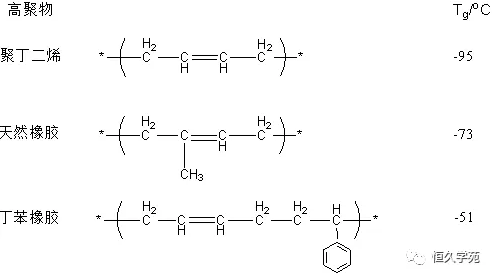
主链中含有孤立双键的高聚物,虽然双键本身不能内旋转,但双键旁的α单键更易旋转,所以Tg都比较低。例如,丁二烯类橡胶都有较低的玻璃化温度。
(2)取代基
旁侧基团的极性,对分子链的内旋转和分子间的相互作用都会产生很大的影响。侧基的极性越强,Tg越高。一些烯烃类聚合物的Tg与取代基极性的关系如表2所示。
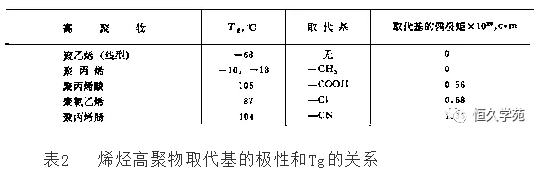
此外,增加分子链上极性基团的数量,也能提高高聚物的Tg.但当极性基团的数量超过一定值后,由于它们之间的静电斥力超过吸引力,反而导致分子链间距离增大,Tg下降。取代基的位阻增加,分子链内旋转受阻碍程度增加,Tg升高。
应当强调指出,侧基的存在并不总是使Tg增大的。取代基在主链上的对称性对Tg也有很大影响,聚偏二氯乙烯中极性取代基对称双取代,偶极抵销一部分,整个分子极性矩减小,内旋转位垒降低,柔性增加,其Tg比聚氯乙烯为低;而聚异丁烯的每个链节上,有两个对称的侧甲基,使主链间距离增大,链间作用力减弱,内旋转位垒降低,柔性增加,其Tg比聚丙烯为低。又如,当高聚物中存在柔性侧基时,随着侧基的增大,在一定范围内,由于柔性侧基使分子间距离加大,相互作用减弱,即产生“内增塑”作用,所以,Tg反而下降。(3)几何异构
单取代烯类高聚物如聚丙烯酸酯、聚苯乙烯等的玻璃化温度几乎与它们的立构无关,而双取代烯类高聚物的玻璃化温度都与立构类型有关。一般,全同立构的Tg较低,间同立构的Tg较高。在顺反异构中,往往反式分子链较硬,Tg较大。
分子链间有离子键可以显著提高Tg。例如,聚丙烯酸中加入金属离子,Tg会大大提高,其效果又随离子的价数而定。用Na+使Tg从l06℃提高到280℃;用Cu2+取代Na+, Tg提高到500℃。
无规共聚物的Tg介于两种共聚组分单体的Tg之间,并且随着共聚组分的变化,其Tg在两种均聚物的Tg之间线性或非线性变化。非无规共聚物中,zui简单的是交替共聚,他们可以看成是两种单体组成一个重复单元的均聚物,因此只有一个Tg。而嵌段或接枝共聚物情况就复杂多了。
(2)交联随着交联点的增加,高聚物自由体积减少,分子链的运动受到约束的程度也增加,相邻交联点之间平均链长变小,所以Tg升高。
分子量的增加使Tg增加,特别是在分子量很小时,这种影响明显,当分子量超过一定的程度后,Tg随分子量变化就不明显了。
增塑剂对Tg的影响也是相当显著的,玻璃化温度较高的聚合物在加入增塑剂后,可以使Tg明显下降。例如:纯的聚氯乙烯Tg=78℃,在室温下是硬塑料,加入45%的增塑剂后,Tg=-30℃,可以作为橡胶代用品。淀粉的玻璃化温度在加水前后就有明显的变化。
马德柱,何平笙等《高举伍的结构和性能》 科学出版社 1995.06
何曼君,陈维孝等《高分子物理》 复旦大学出版社 2004.06
于同隐,唐敖庆等《高分子化学与物理专论》 中山大学出版社 1984.07
任金霞,曹益林等. 利用神经网络预测聚合物玻璃化温度,河南科学,1999,17(3).
Walter Kauzmann, The nature of the glassy state and the behavior ofliquids at low temperatures
文章来源:来源于互联网,如有侵权请作者联系我们!


免责声明:非本网作品均来自互联网,发布目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。如涉及作品内容、版权和其他问题,请及时与本网联系,我们将核实后进行删除,本网站对此声明具有最终解释权。